PIPELINE &
INNOVATION
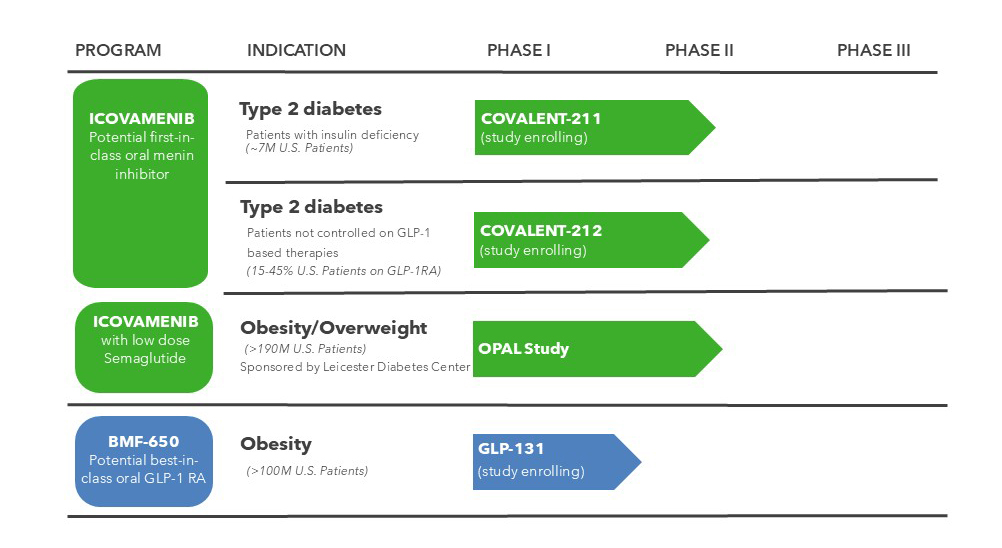
Pipeline
Our Science
Biomea Fusion is developing a new generation of small molecules that target metabolic diseases with high unmet need.
Icovamenib
A Breakthrough in Diabetes Innovation
Icovamenib is a first-in-class therapy with a novel mechanism of action, uniquely designed to address the underlying causes of diabetes. Unlike conventional treatments, it delivers a durable treatment effect by improving beta cell function and enhancing the incretin effect, helping to restore the body’s natural ability to regulate blood sugar.
This innovative therapy is administered as a simple, oral, once-daily treatment over a 12-week period, offering a convenient and patient-friendly approach. Icovamenib has been shown to enhance endogenous insulin production and improve beta cell function*, supporting long-term metabolic health.
Beyond glucose control, icovamenib has demonstrated additional metabolic benefits, including promoting body weight loss* and increasing the proportion of lean mass while preserving existing lean mass in preclinical studies. These effects contribute to improved overall health and disease management.
Current Clinical Trials
- COVALENT-112 – Phase II Trial of Icovamenib in Participants with Type 1 Diabetes Mellitus – Study Ongoing
- COVALENT-211 – Phase II Trial of Icovamenib in Participants with Insulin-Deficient Type 2 Diabetes – Study Enrolling
- COVALENT-212 – Phase II Trial of Icovamenib in Participants with Type 2 Diabetes Uncontrolled with a GLP-1 RA-based Therapy – Study Enrolling
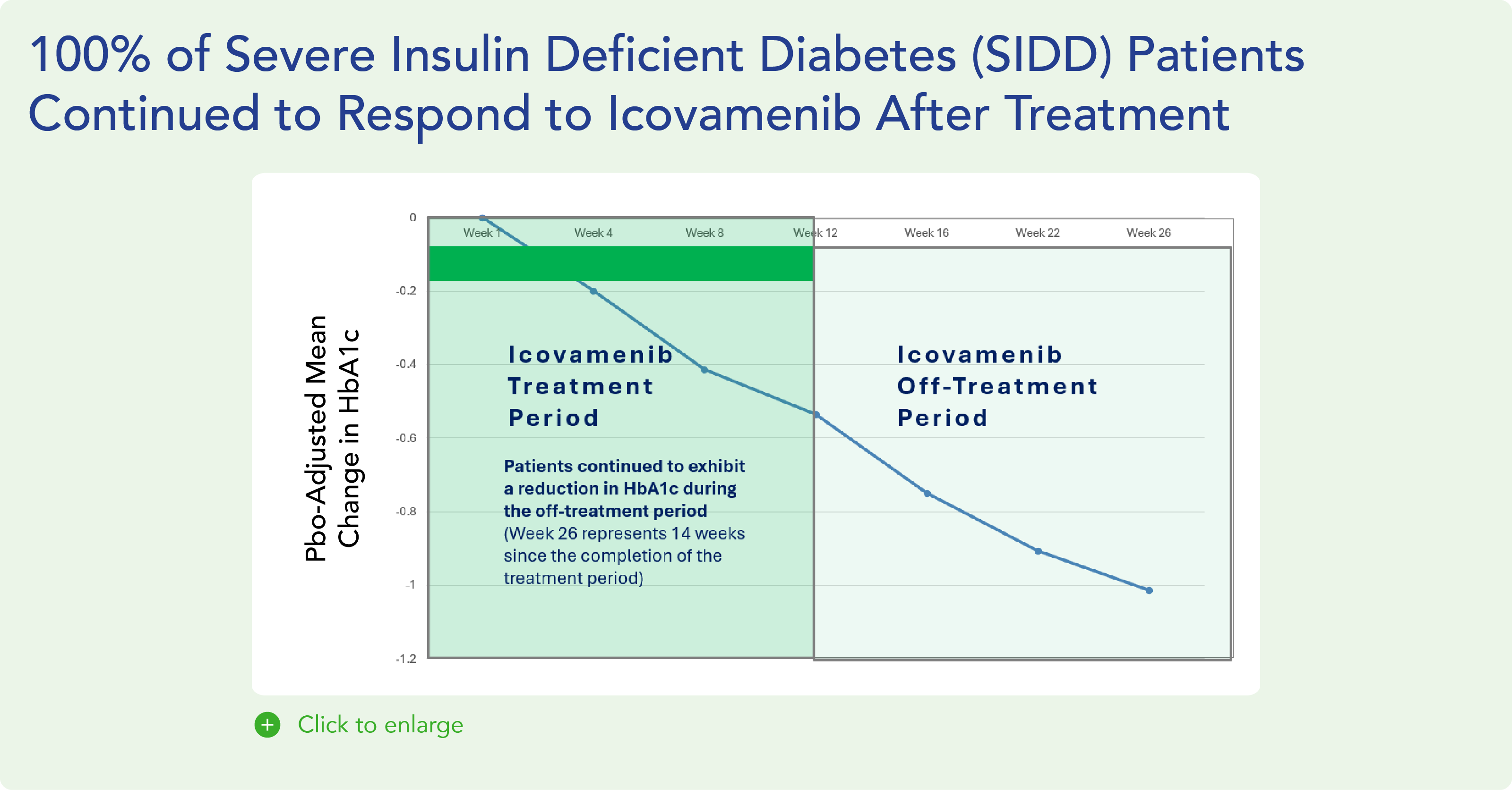
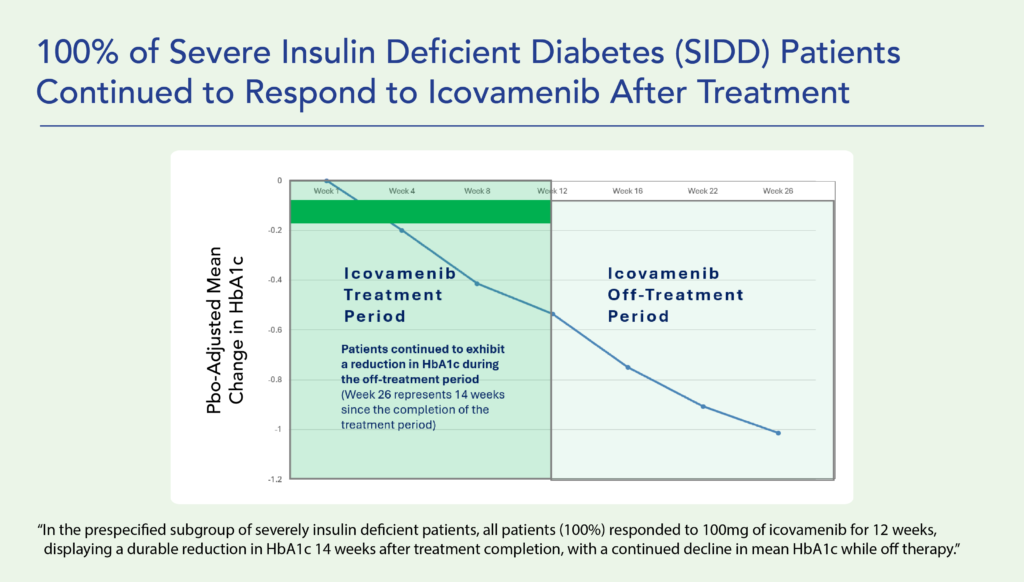
Development Rationale
Research shows that individuals with the lowest insulin production face the highest all-cause mortality and the greatest risk of treatment failure. Icovamenib has demonstrated superior treatment efficacy in this vulnerable patient population, offering a promising solution for those with the most urgent need.
Furthermore, icovamenib has been shown to promote weight loss and increase muscle mass percentage when combined with a GLP-1 receptor agonist (RA) therapy*. This supports its potential use in combination with GLP-1-based therapies, including as a treatment following the failure of a GLP-1-based therapy.
*Clinical results are based on preliminary findings.
BMF-650
A Next-Generation Oral Small Molecule GLP-1 Receptor Agonist
Biomea Fusion is prioritizing the development of BMF-650, an investigational, next-generation, oral small-molecule GLP-1 receptor agonist (GLP-1 RA), which has demonstrated positive, early preclinical activity, including improved glucose-stimulated insulin secretion, reduction in blood glucose concentration, appetite suppression and weight loss in cynomolgus monkeys.
BMF-650 is currently being evaluated in a Phase I clinical study in healthy participants.